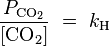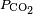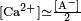Calcium carbonate is poorly soluble in pure water (47 mg/L at normal atmospheric CO2 partial pressure as shown below).
The equilibrium of its solution is given by the equation (with dissolved calcium carbonate on the right):
CaCO3
Ca2+ + CO32−
Ksp = 3.7×10−9 to 8.7×10−9at 25 °C
where the
solubility product for [Ca2+][CO32−] is given as anywhere from
Ksp = 3.7×10−9 to
Ksp = 8.7×10−9 at 25 °C, depending upon the data source.
[41][42] What the equation means is that the product of molar concentration of calcium ions (
moles of dissolved Ca2+ per liter of solution) with the molar concentration of dissolved CO32− cannot exceed the value of
Ksp. This seemingly simple solubility equation, however, must be taken along with the more complicated equilibrium of
carbon dioxide with
water (see
carbonic acid). Some of the CO32− combines with H+ in the solution according to:
HCO3−
H+ + CO32−
Ka2 = 5.61×10−11 at 25 °C
HCO3− is known as the
bicarbonate ion.
Calcium bicarbonate is many times more soluble in water than calcium carbonate—indeed it exists
only in solution.
Some of the HCO3− combines with H+ in solution according to:
H2CO3
H+ + HCO3−
Ka1 = 2.5×10−4 at 25 °C
Some of the H2CO3 breaks up into water and dissolved carbon dioxide according to:
H2O + CO2(dissolved)
H2CO3
Kh = 1.70×10−3 at 25 °C
And dissolved carbon dioxide is in equilibrium with atmospheric carbon dioxide according to:
where
kH = 29.76 atm/(mol/L) at 25 °C (
Henry constant),
being the CO2 partial pressure.
For ambient air,
is around 3.5×10−4 atmospheres (or equivalently 35
Pa). The last equation above fixes the concentration of dissolved CO2 as a function of
, independent of the concentration of dissolved CaCO3. At atmospheric partial pressure of CO2, dissolved CO2 concentration is 1.2×10−5 moles/liter. The equation before that fixes the concentration of H2CO3 as a function of [CO2]. For [CO2]=1.2×10−5, it results in [H2CO3]=2.0×10−8 moles per liter. When [H2CO3] is known, the remaining three equations together with
Calcium ion solubility as a function of
CO2 partial pressure at 25 °C (Ksp = 4.47×10−9)
(atm) pH [Ca2+] (mol/L)
10−12 12.0 5.19 × 10−3
10−10 11.3 1.12 × 10−3
10−8 10.7 2.55 × 10−4
10−6 9.83 1.20 × 10−4
10−4 8.62 3.16 × 10−4
3.5 × 10−4 8.27 4.70 × 10−4
10−3 7.96 6.62 × 10−4
10−2 7.30 1.42 × 10−3
10−1 6.63 3.05 × 10−3
1 5.96 6.58 × 10−3
10 5.30 1.42 × 10−2
H2O
H+ + OH−
K = 10−14 at 25 °C
(which is true for all aqueous solutions), and the fact that the solution must be electrically neutral,
2[Ca2+] + 2[H+] = [HCO3−] + 2[CO32−] + [OH−]
make it possible to solve simultaneously for the remaining five unknown concentrations (note that the above form of the neutrality equation is valid only if calcium carbonate has been put in contact with pure water or with a neutral pH solution; in the case where the origin water solvent pH is not neutral, the equation is modified).
The table on the right shows the result for [Ca2+] and [H+] (in the form of pH) as a function of ambient partial pressure of CO2 (
Ksp = 4.47×10−9 has been taken for the calculation).
- At atmospheric levels of ambient CO2 the table indicates the solution will be slightly alkaline with a maximum CaCO3 solubility of 47 mg/L.
- As ambient CO2 partial pressure is reduced below atmospheric levels, the solution becomes more and more alkaline. At extremely low
, dissolved CO2, bicarbonate ion, and carbonate ion largely evaporate from the solution, leaving a highly alkaline solution of calcium hydroxide, which is more soluble than CaCO3. Note that for
= 10−12 atm, the [Ca2+][OH−]2 product is still below the solubility product of Ca(OH)2 (8×10−6). For still lower CO2 pressure, Ca(OH)2 precipitation will occur before CaCO3 precipitation.
- As ambient CO2 partial pressure increases to levels above atmospheric, pH drops, and much of the carbonate ion is converted to bicarbonate ion, which results in higher solubility of Ca2+.
The effect of the latter is especially evident in day-to-day life of people who have hard water. Water in aquifers underground can be exposed to levels of CO2 much higher than atmospheric. As such water percolates through calcium carbonate rock, the CaCO3dissolves according to the second trend. When that same water then emerges from the tap, in time it comes into equilibrium with CO2levels in the air by outgassing its excess CO2. The calcium carbonate becomes less soluble as a result and the excess precipitates as lime scale. This same process is responsible for the formation of
stalactites and
stalagmites in limestone caves.
Two hydrated phases of calcium carbonate,
monohydrocalcite, CaCO3·H2O and
ikaite, CaCO3·6H2O, may
precipitate from water at ambient conditions and persist as metastable phases.
With varying pH, temperature and salinity: CaCO3 scaling in swimming pools[edit]


In contrast to the open equilibrium scenario above, many swimming pools are managed by addition of
sodium bicarbonate (NaHCO3) to about 2 mM as a buffer, then control of pH through use of HCl, NaHSO4, Na2CO3, NaOH or chlorine formulations that are acidic or basic. In this situation, dissolved inorganic carbon (
DIC) is far from equilibrium with atmospheric CO2. Progress towards equilibrium through outgassing of CO2 is slowed by (i) the slow reaction
H2CO3 ⇌ CO2(aq) + H2O;
[43] (ii) limited aeration in a deep water column and (iii) periodic replenishment of bicarbonate to maintain buffer capacity (often estimated through measurement of
‘total alkalinity’).
In this situation, the dissociation constants for the much faster reactions H2CO3 ⇌ H+ + HCO3‾ ⇌ 2 H+ + CO32− allow the prediction of concentrations of each DIC species in solution, from the added concentration of HCO3− (which comprises more than 90% of
total DIC from pH 7 to pH 8 at 25 ˚C in fresh water.
[44] Addition of HCO3− will increase CO32− concentration at any pH. Rearranging the equations given above, we can see that [Ca2+] = Ksp / [CO32−], and [CO32−] = Ka2 × [HCO3−] / [H+]. Therefore, when HCO3− concentration is known, the maximum concentration of Ca2+ ions before scaling through CaCO3 precipitation can be predicted from the formula:
Ca2+max = (Ksp / Ka2) × ([H+] / [HCO3−])
The solubility product for CaCO3 (Ksp) and the dissociation constants for the DIC species (including Ka2) are all substantially affected by temperature and
salinity,
[44] with the overall effect that Ca2+max increases from fresh to salt water, and decreases with rising temperature, pH, or added bicarbonate level, as illustrated in the accompanying graphs.
The trends are illustrative for pool management, but whether scaling occurs also depends on other factors including interactions with Mg2+, B(OH)4− and other ions in the pool, as well as supersaturation effects.
[45][46] Scaling is commonly observed in electrolytic chlorine generators, where there is a high pH near the cathode surface and scale deposition further increases temperature. This is one reason that some pool operators prefer borate over bicarbonate as the primary pH buffer, and avoid the use of pool chemicals containing calcium.
[47]
Solubility in a strong or weak acid solution[edit]
Solutions of
strong (
HCl), moderately strong (
sulfamic) or
weak (
acetic,
citric,
sorbic,
lactic,
phosphoric) acids are commercially available. They are commonly used as
descaling agents to remove
limescale deposits. The maximum amount of CaCO3 that can be "dissolved" by one liter of an acid solution can be calculated using the above equilibrium equations.
- In the case of a strong monoacid with decreasing acid concentration [A] = [A−], we obtain (with CaCO3 molar mass = 100 g):
[A] (mol/L) 1 10−1 10−2 10−3 10−4 10−5 10−6 10−7 10−10
Initial pH 0.00 1.00 2.00 3.00 4.00 5.00 6.00 6.79 7.00
Final pH 6.75 7.25 7.75 8.14 8.25 8.26 8.26 8.26 8.27
Dissolved CaCO3(g per liter of acid) 50.0 5.00 0.514 0.0849 0.0504 0.0474 0.0471 0.0470 0.0470
where the initial state is the acid solution with no Ca2+ (not taking into account possible CO2 dissolution) and the final state is the solution with saturated Ca2+. For strong acid concentrations, all species have a negligible concentration in the final state with respect to Ca2+ and A− so that the neutrality equation reduces approximately to 2[Ca2+] = [A−] yielding
. When the concentration decreases, [HCO3−] becomes non-negligible so that the preceding expression is no longer valid. For vanishing acid concentrations, one can recover the final pH and the solubility of CaCO3 in pure water.
- In the case of a weak monoacid (here we take acetic acid with pKA = 4.76) with decreasing total acid concentration [A] = [A−]+[AH], we obtain:
[A] (mol/L) 1 10−1 10−2 10−3 10−4 10−5 10−6 10−7 10−10
Initial pH 2.38 2.88 3.39 3.91 4.47 5.15 6.02 6.79 7.00
Final pH 6.75 7.25 7.75 8.14 8.25 8.26 8.26 8.26 8.27
Dissolved CaCO3(g per liter of acid) 49.5 4.99 0.513 0.0848 0.0504 0.0474 0.0471 0.0470 0.0470
For the same total acid concentration, the initial pH of the weak acid is less acid than the one of the strong acid; however, the maximum amount of CaCO3 which can be dissolved is approximately the same. This is because in the final state, the pH is larger than the p
KA, so that the weak acid is almost completely dissociated, yielding in the end as many H+ ions as the strong acid to "dissolve" the calcium carbonate.
- The calculation in the case of phosphoric acid (which is the most widely used for domestic applications) is more complicated since the concentrations of the four dissociation states corresponding to this acid must be calculated together with [HCO3−], [CO32−], [Ca2+], [H+] and [OH−]. The system may be reduced to a seventh degree equation for [H+] the numerical solution of which gives
[A] (mol/L) 1 10−1 10−2 10−3 10−4 10−5 10−6 10−7 10−10
Initial pH 1.08 1.62 2.25 3.05 4.01 5.00 5.97 6.74 7.00
Final pH 6.71 7.17 7.63 8.06 8.24 8.26 8.26 8.26 8.27
Dissolved CaCO3(g per liter of acid) 62.0 7.39 0.874 0.123 0.0536 0.0477 0.0471 0.0471 0.0470
where [A] = [H3PO4] + [H2PO4−] + [HPO42−] + [PO43−] is the total acid concentration. Thus phosphoric acid is more efficient than a monoacid since at the final almost neutral pH, the second dissociated state concentration [HPO42−] is not negligible (see
phosphoric acid).